Jetzt Kontakt zu uns aufnehmen!
Sie erreichen uns unter folgender Telefonnummer:
+49 (0) 385 477 419 00




Hersteller von Medizinprodukten sind verpflichtet, EN ISO 17664 konforme Gebrauchsanweisungen mitzuliefern. Diese Anweisungen müssen validierte Aufbereitungsprozesse für das jeweilige Medizinprodukt enthalten. HygCen ist ein akkreditiertes Prüflabor und prüft die in Ihrer Gebrauchsanweisung genannten Aufbereitungsprozesse gemäß EN ISO 17664 und alle damit verbundenen Standards (z. B. EN ISO 15883 Reihe - Reinigungs-Desinfektionsgeräte, EN ISO 17665 - Sterilisatoren oder EN ISO 10993 Reihe zur Biokompatibilität).
In Deutschland gibt es jährlich 1 Million Krankenhausinfektionen. Wir sagen: Das sind 1 Million zu viel. Mit EN ISO 17664 konformen Gebrauchsanweisungen schützen Sie Patient*innen und Anwender*innen.
Beziehen Sie HygCen frühzeitig in die Prüfung Ihrer Aufbereitungsprozesse Ihrer Produkte für die Gesundheitsfürsorge mit ein. So können Sie sich als Hersteller absichern. Vermeiden Sie unnötige Risiken und Kosten!
Aufbereitete Medizinprodukte müssen für Patient*innen genauso sicher sein wie erst- oder einmalig verwendete. Das heißt: Die Produkte dürfen weder mikrobiell kontaminiert noch funktional beeinträchtigt sein. Als Hersteller sind Sie verpflichtet, ein validiertes Aufbereitungsverfahren zu empfehlen. Dieses garantiert, dass das Medizinprodukt bei erneuter Anwendung frei von jeglichen lebensfähigen Mikroorganismen ist.
Die Aufbereitung von Medizinprodukten fordert jede*n verantwortliche*n Aufbereiter*in und Anwender*in. Der Gesetzgeber verpflichtet die Hersteller, einen optimalen Aufbereitungsprozess für jedes Medizinprodukt anzugeben. HygCen validiert diese Herstellerangaben gemäß EN ISO 17664.
Die Prüfung gewährleistet, dass das Medizinprodukt mithilfe der Herstellerangaben wirksam aufbereitet werden kann. Es gilt einzelne Arbeitsschritte, ihre Wechselwirkungen zueinander und das absehbare Ergebnis des Aufbereitungsprozesses zu berücksichtigen.
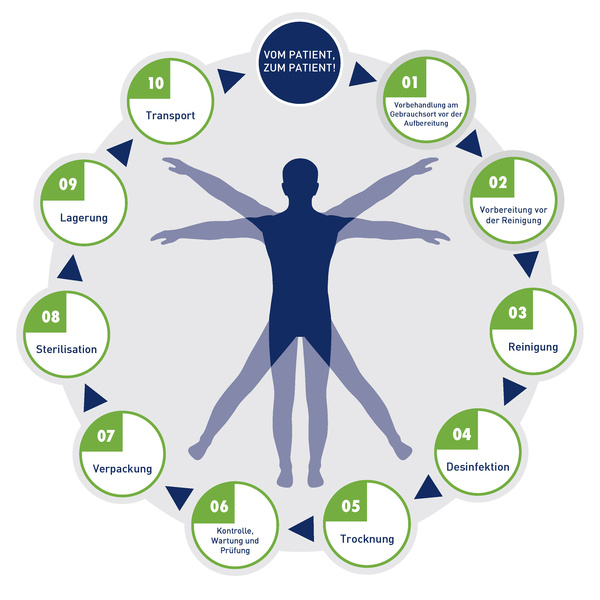
Hersteller von Medizinprodukten müssen konkrete Aufbereitungsinformationen angeben. Die EN ISO 17664 legt die Anforderungen dafür fest. Möchten Sie ein neues Produkt auf dem europäischen bzw. internationalen Markt einführen, dient Ihnen dieser internationale Normenstandard als wichtige Hilfestellung. Der Aufbereitungszyklus von Medizinprodukten wird gemäß EN ISO 17664 wie folgt unterteilt:
Wenn Sie als Hersteller so detaillierte Angaben machen, vermutet der Gesetzesgeber, dass Sie geltendes Recht (EU Medical Device Regulation MDR) erfüllen.
Pflichten als Aufbereiter*in gemäß EN ISO 17664
Aufbereiter von Medizinprodukten unterliegen ebenfalls gesetzlichen Pflichten. Sie müssen das Medizinprodukt nicht nur reinigen, desinfizieren und ggf. sterilisieren. Der Aufbereiter prüft auch die technisch-funktionelle Sicherheit bzw. stellt diese wieder her. Um das zu gewährleisten, müssen die Aufbereiter*innen laut Verordnung,
Eine ordnungsgemäße Aufbereitung wird vermutet, wenn der Aufbereiter gemäß den RKI-BfArM-Empfehlungen* arbeitet. Weiterhin unterliegt die Aufbereitung den anerkannten Regeln der Technik und den Arbeitsschutz- und Unfallverhütungsvorschriften.
*RKI-BfArM-Empfehlungen: Gemeinsame Empfehlungen der Kommission für Krankenhaushygiene und Infektionsprävention beim Robert Koch-Institut und des Bundesinstitutes für Arzneimittel und Medizinprodukte.
Einkäufer*innen von Medizinprodukten unterliegen einer hohen Verantwortung.
Sie sollten sich mit den Anwender*innen und Aufbereiter*innen abstimmen und folgende Fragen prüfen:
Ist die Aufbereitungsanweisung mangelhaft, muss der Einkäufer beim Hersteller eine EN ISO 17664 konforme Gebrauchsanweisung erfragen. Falls der Hersteller daraufhin keine liefert, verstößt er ggf. gegen das Medizinprodukterecht. Zum Beispiel, wenn das Produkt schon lange im Verkehr ist.
Informieren Sie als Einkäufer*in die zuständige Überwachungsbehörde, um selbst einem Verstoß vorzubeugen. Hat die fehlende Gebrauchsanweisung bereits zu einem Vorkommnis* geführt, muss das beim Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) gemeldet werden (gem. Medizinproduktesicherheitsplanverordnung).
*Ein „Vorkommnis“ hat sich ereignet, wenn ein*e Patient*in, Anwender*in oder ein Dritter aufgrund einer mangelnden Gebrauchsanweisung zu Schaden gekommen ist.
HygCen validiert Herstellerangaben in Gebrauchsanweisungen gemäß EN ISO 17664. Die harmonisierte Norm EN ISO 17664 zur Sterilisation von Medizinprodukten trägt seit 2018 den neuen Titel „Aufbereitung von Produkten für die Gesundheitsfürsorge – Vom Medizinprodukt-Hersteller bereitzustellende Informationen für die Aufbereitung von Medizinprodukten“. Mit der Umbenennung gehen zusätzliche Pflichten für Hersteller, Einkäufer*innen und Aufbereiter*innen einher. Generell der Fokus auf die Validierung des Aufbereitungsprozesses erhöht.
Medizinprodukte, die in den USA zugelassen werden sollen, müssen die die Anforderungen der Food and Drug Administration (FDA) erfüllen. HygCen prüft Ihr Aufbereitungsprozesse auch auf FDA-Konformität. Dafür muss das Prüflabor einen individuellen Validierungsplan erstellen. Bitte beauftragen Sie diesen separat zur Prüfung gemäß EN ISO 17664.
Als Hersteller sind Sie verpflichtet, alle notwendigen Informationen zur Wiederaufbereitung anzugeben. Aufbereiter sollen anhand der Gebrauchsanweisung Medizinprodukte vor jedem Einsatz sachgerecht aufbereiten können. Die EN ISO 17664 Richtlinie legt fest, wie detailliert das Aufbereitungsverfahren in der Gebrauchsanweisung beschrieben werden muss. HygCen validiert Ihre Gebrauchsanweisung gemäß EN ISO 17664 und ggf. weiterer Standards, wie z. B. EN ISO 10993 Reihe zur Biokompatibilität. Dabei prüfen wir alle vorhandenen Aufbereitungsverfahren. Zum Beispiel die manuelle Aufbereitung durch Reinigung und Desinfektion, die maschinelle Aufbereitung (thermisch) und verschiedene Sterilisationsverfahren (Dampf, H2O2, Ethylenoxid (EO), Formaldehyd etc.).
Gerne erstellen wir für die Prüfung der Aufbereitungsprozesse Ihrer Gebrauchsanweisung einen individuellen Validierungsplan!
Seit Mitte 2017 gilt die überarbeitete Medizinproduktebetreiberverordnung (MPBetreibV). Die Verordnung regelt das Errichten, Betreiben und Anwenden von Medizinprodukten. Die aktuell gültige MPBetreibV umfasst jetzt 19 statt 15 Paragrafen. Die Verordnung basiert auf dem Medizinproduktgesetz (MPG). Danach ist genau festgelegt, was Medizinprodukte sind und was nicht (z. B. Arzneimittel und Kosmetikprodukte). Im medizinischen Alltag sind vor allem (wiederaufbereitbare) Instrumente Medizinprodukte im Sinne des MPGs. Diese werden je nach Verwendung und Risiko für die Patienten*innen gemäß Richtlinie 93/42/EWG klassifiziert. Für die Aufbereitung werden Medizinprodukte zusätzlich gemäß den Vorgaben der RKI-BfArM-Empfehlung klassifiziert. Wird ein Instrument als semi-kritisch oder kritisch eingestuft, kann das erhebliche Konsequenzen für die Qualität und Dokumentation der Aufbereitungsverfahren haben.
Seit 2002 regelt die MPBetreibV die Pflichten der Betreiber und Anwender von Medizinprodukten.
Laut Verordnung darf z. B. nur eingewiesenes Personal Medizinprodukte anwenden. Werden Medizinprodukte wiederholt eingesetzt, müssen diese immer korrekt funktionieren und korrekt aufbereitet sein. Für aktive Medizinprodukte legt die MPBetreibV weitere Rahmenbedingungen fest. Dazu gehören die Bereitstellung einer geeigneten Infrastruktur sowie der Aufbau eines Dokumentationssystems.
Für die Medizinproduktebetreiberverordnung traten 2014 und 2017 wichtige Änderungen in Kraft.
Der Begriff des „Betreibers" wird präzisiert (§ 2 (2)).Betreiber von Medizinprodukten sind diejenigen, die für den Betrieb in einer Gesundheitseinrichtung verantwortlich sind. Dazu gehören auch selbstständige Ärzt*innen (z. B. Narkoseärzt*innen) oder Physiotherapeut*innen. Wer bspw. Defibrillatoren öffentlich zugänglich bereitstellt (auch außerhalb von medizinischen Einrichtungen), ist ebenfalls Betreiber. Hersteller und Aufbereiter sind dagegen keine Betreiber.
Alle Personen, die Tätigkeiten ausführen, die der MPBetreibV unterliegen, müssen folgende Anforderungen erfüllen (§ 5).Die Personen müssen über aktuelle Fachkenntnisse und -qualifikationen verfügen. Sie dürfen hinsichtlich der Beurteilung keiner Weisung unterliegen. Zudem müssen sie über alle Mittel verfügen, die zur ordnungsgemäßen Ausübung ihrer Tätigkeit notwendig sind.
Betriebe ab 20 Mitarbeiter*innen benötigen eine*n Beauftragte*n für Medizinproduktsicherheit (§ 6). Als zentrale*r Ansprechpartner*in verantwortet sie/er die Umsetzung der MPBetreibV. Die/der Beauftragte kommuniziert mit Behörden, Herstellern und Vertreibern. Sie/er meldet Risiken und leitet ggf. Korrekturmaßnahmen ein. Zudem koordiniert sie/er alle internen Prozesse, die zur Erfüllung von Melde- und Mitwirkungspflichten der Anwender*innen und Betreiber dienen. Die/der Beauftragte leitet ebenfalls korrektive Maßnahmen, wie z. B. Rückrufmaßnahmen.
Die/der Beauftragte für Medizinproduktsicherheit muss erreichbar sein. Zu diesem Zweck ist ihre/seine E-Mail-Adresse auf der Internetseite der Gesundheitseinrichtung zu veröffentlichen. Die/der Beauftragte darf außerdem nicht bei seiner Aufgabenerfüllung behindert werden.
Basierend auf der novellierten Medizinproduktebetreiberverordnung sind zukünftig weitere Anforderungen wichtig. Für das Errichten, Bereithalten, Instandhalten, Aufbereiten sowie der sicherheits- und messtechnischen Kontrollen gelten u.a. folgende (neue) Pflichten:
Beim Führen des Medizinproduktebuchs ist der Betreiber zu folgendem verpflichtet:

Die neue Medical Device Regulation hat die bisherigen Medizinprodukte-Richtlinien 2017 abgelöst. Bisher galt für Medizinprodukte die Medical Device Directive (Richtlinie 93/42/EWG) und für aktive implantierbare Medizinprodukte die Active Implantable Medical Device (Richtlinie 90/385/EWG).
Die MDR ist gekennzeichnet durch einen deutlich höherer Dokumentationsaufwand für die Hersteller. Das betrifft speziell Produkte mit erhöhtem Risiko. Marktüberwachungsbehörden können gemäß MDR feststellen, ob Produkte rechtskonform sind. Wurde keine EU-Konformitätserklärung erstellt, ist die Erklärung unvollständig oder technisch nicht dokumentierbar, kann das Medizinprodukt als amtlich nichtkonform bezeichnet werden. Stellt der Hersteller die Konformität innerhalb der Fristen nicht wieder her, kann dem Medizinprodukt der Marktzugang untersagt werden.
In der Medical Device Regulation werden zwei eigenständige Medizinprodukte-Richtlinien zusammengefasst. Darüber hinaus konkretisiert die MDR bestimmte Anforderungen:
Die EN ISO 17664-Normenreihe legt fest, welche Informationen Hersteller für die Aufbereitung (Reinigung, Desinfektion, ggf. Sterilisation) von Medizinprodukten bereitstellen müssen. Ziel ist die sichere Wiederverwendung und die Minimierung von Infektionsrisiken.
Die Normenreihe gilt für wiederverwendbare Medizinprodukte, die gereinigt, desinfiziert und/oder sterilisiert werden müssen, bevor sie erneut eingesetzt werden. Teil 1 betrifft Produkte mit Kontakt zu sterilen Körperbereichen, Schleimhäuten oder veränderter Haut; Teil 2 unkritische Produkte mit Kontakt zu intakter Haut oder ohne Patientenkontakt.
Hersteller müssen detaillierte, validierte Anweisungen zu allen Schritten der Aufbereitung liefern: Vorbehandlung, Reinigung, Desinfektion, ggf. Sterilisation, Trocknung, Kontrolle, Verpackung, Lagerung und Transport.
Nur validierte Aufbereitungsverfahren gewährleisten, dass Medizinprodukte sicher und wirksam gereinigt, desinfiziert und/oder sterilisiert werden. Die Validierung ist Voraussetzung für die Konformität mit der EN ISO 17664-Normenreihe und regulatorischen Vorgaben.
HygCen bietet umfassende Beratung, Prüfung und Validierung Ihrer Aufbereitungsanweisungen nach EN ISO 17664-1 und -2. Wir begleiten Sie von der Risikoanalyse bis zur Dokumentation und unterstützen bei Audits und Behördenanfragen.
Sie erreichen uns unter folgender Telefonnummer:
+49 (0) 385 477 419 00
Sie finden uns hier:
HygCen Germany GmbH
Bornhövedstrasse 78
19055 Schwerin