


HygCen examines the reprocessing processes of medical devices
Manufacturers of medical devices are required to provide EN ISO 17664 compliant instructions for use. These instructions must contain validated reprocessing processes for the respective medical device. HygCen is an accredited testing laboratory and we test your reprocessing processes stated in your instructions for use according to EN ISO 17664 and all related standards (eg EN ISO 15883 series - washer-disinfectors, EN ISO 17665 sterilizers or EN ISO 10993 series on biocompatibility). There are 1 million hospital infections annually in Germany. We say: That's 1 million too much. With EN ISO 17664 compliant instructions you protect patients and users. Involve HygCen at an early stage in the review of your healthcare reprocessing process. So you can hedge yourself as a manufacturer. Avoid unnecessary risks and costs!
Recycled medical devices must be as safe for the patient as first or single used.This means that the products must not be microbially contaminated or functionally impaired. As a manufacturer, you are required to recommend a validated reprocessing process.This ensures that the medical device is free from any viable microorganisms when reused.
What are suitable validated reprocessing processes?
The reprocessing of medical devices requires every responsible preparer and user. Legislation requires manufacturers to specify an optimal reprocessing process for each medical device. HygCen validates these instructions for use in accordance with EN ISO 17664. The testing laboratory ensures that the medical device can be effectively processed using the manufacturer's instructions. It is necessary to consider individual work steps, their interactions with each other and the foreseeable sterility level.
Your obligation with regards to reprocess medical devices
- State a reprocessing process
As a manufacturer, you must state one reprocessing process. - Method of detection
As a manufacturer, you must ensure a valid reprocessing process by demonstrating the cleaning an disinfection efficacy. - Criteria for efficacy
As an manufacturer, you must specify how and often your medical device can be effectively processed by the user.
HygCen validates your instructions for use according to EN ISO 17664
- Safe reprocessing process
HygCen validates your stated process. Only then you as a manufacturer may recommend it. - Prove of patients safety
HygCen determines verifiably the cleaning and disinfection efficacy. - Duration of use
HygCen checks how often your medical device can be processed without damage.
What should the reprocessing instructions look like?
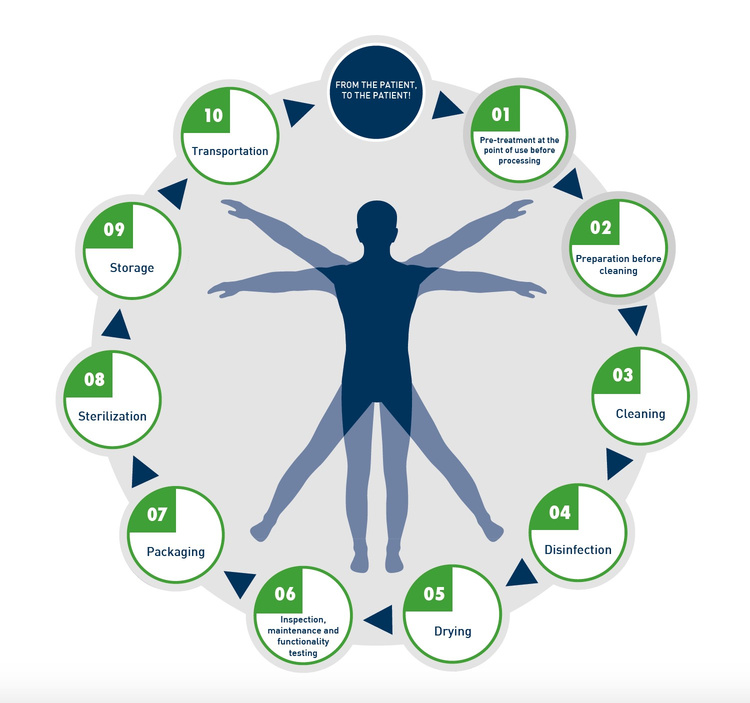
Manufacturers of medical devices must provide specific reprocessing information. EN ISO 17664 specifies the requirements for this. If you would like to introduce a new product on the European or international market, this international standard will serve as an important aid. The reprocessing cycle of medical devices is subdivided according to EN ISO 17664 as follows:
If you provide such detailed information as the manufacturer, the legislator assumes that you comply with EU Medical Device Regulation (MDR).
Preparers of medical devices are also subject to legal obligations. Not only do you need to clean, disinfect and, if necessary, sterilize the medical device. The preparer also checks the technical-functional safety or restores it. To ensure this, the preparers have to:
- Follow the manufacturer's instructions for reprocessing. (MPBetreibV of 2016: § 8, par.1)
- Carry out the reprocessing with validated procedures. This must be retracable and ensure that neither patient, user or third parties are at risk.
- Be qualified for the reprocessing of medical devices. (MPBetreibV of 2016: § 5, sentence 1
- Have basic equipment that meets the defined requirements. (MPBetreibV of 2016: § 5, sentence 3)
- Proper reprocessing is suspected if the conditioner works in accordance with the RKI BfArM recommendations *. Furthermore, the reprocessing is subject to the recognized rules of technology and the occupational safety and accident prevention regulations.
* RKI-BfArM Recommendations: Joint Recommendations of the Commission for Hospital Hygiene and Infection Prevention at the Robert Koch Institute and the Federal Institute for Drugs and Medical Devices.
Purchasers of medical devices are subject to a high degree of responsibility. You should coordinate with the users and preparers and check the following questions:
1. Does the instructions for use meet the legal requirements?
2. Can the prescribed reprocessing process be technically and personally implemented by the operator?
If the reprocessing instruction is defective, the purchaser must ask the manufacturer for an EN ISO 17664 compliant user manual. If the manufacturer does not deliver then, it may violate the medical device law. For example, if the product has been in circulation for a long time.
As a purchaser, inform the relevant monitoring authority to prevent yourself from being infringed. If the missing instructions for use have already led to an incident *, this must be reported to the Federal Institute for Drugs and Medical Devices (BfArM) (in accordance with the Medical Product Safety Plan Ordinance).
* An "incident" has occurred when a patient, user or a third party has been harmed due to a lack of instructions for use.
Instructions for use acc. EN ISO 17664
EN ISO 17664 compliant instructions for use
HygCen validates the manufacturer's instructions for use in accordance with EN ISO 17664. The harmonized standard EN ISO 17664 for the sterilization of medical devices has been bearing the new title "Preparing healthcare products - Information to be provided by the medical device manufacturer for the reprocessing of medical devices" since 2018. The renaming implies additional obligations for manufacturers, purchasers and preparers. In general, the validation of the reprocessing process becomes more important.
Medical devices to be approved in the US must meet the requirements of the Food and Drug Administration (FDA). HygCen also checks your product for FDA compliance. For this, the testing laboratory has to create an individual validation plan. Please order this separately for testing in accordance with EN ISO 17664.
As a manufacturer, you are required to provide all necessary information for reprocessing. Staff members should be able to properly reprocess medical devices before each use using the instructions for use. EN ISO 17664 specifies how detailed the reprocessing process must be described in the instructions for use. HygCen validates your instructions for use according to EN ISO 17664 compliance and other standards, if applicable (eg EN ISO 10993 series on biocompatibility). In doing so, we check all available reprocessing processes. For example, the manual reprocessing by cleaning and disinfection, the mechanical reprocessing (thermal) and various sterilization processes (steam, H2O2, ethylene oxide (EO), formaldehyde, etc.).
We will establish an individual validation plan for the reprocessing process of your instructions for use!
We check your instructions according to EN ISO 17664
- Request a quote from us - by phone or via online request.Please tell us for this, which medical device and application area it is. Is there already a reprocessing manual? And what type of reprocessing (manual, mechanical or sterilizing procedures) should we test?
- You send us your product including instructions for use, as soon as you have accepted our offer.
- Immediately after receipt of the goods, we will initiate the testing of the specified reprocessing procedure for your medical device. We will be happy to validate your reprocessing process even before it is included in the instructions for use.
- We test your instructions for use in accordance with EN ISO 17664. Depending on the reprocessing process, we are bound by other standards. For example, the following guidelines: EN ISO 15883 series (washer-disinfectors), EN ISO 17665 (sterilizers) or EN ISO 10993 series (biocompatibility).
- You will receive a digital test report including a professional assessment, secured with a Qualified Electronic Signature (QES). This guarantees the legal validity, integrity, and immutability of your documents – recognized across the world. Our scientific director classifies the results of the test report for you in this assessment. The classification includes an overall assessment and, if applicable, a rating in accordance with EN ISO 17664 as well as other standards and acceptance criteria.
Revised medical device operator regulation
Since mid-2017, the revised Medical Device Operator Ordinance (MPBetreibV) has been in force. The regulation regulates the construction, operation and use of medical devices. The currently valid MPBetreibV now includes 19 instead of 15 paragraphs. The regulation is based on the Medical Devices Act (MPG). Thereafter, it is precisely defined what medical devices are and are not (for example, medicines and cosmetics). In medical practice, (reprocessable) instruments are medical devices in the sense of the MPG. These are classified according to the use and risk to the patient in accordance with Directive 93/42 / EWG. For reprocessing, medical devices are additionally classified according to the specifications of the RKI BfArM Recommendation. If an instrument is classified as semi-critical or critical, this can have significant consequencesfor the quality and documentation of the reprocessing processes.
Since 2002, the MPBetreibV regulates the obligations of the operators and users of medical devices.
According to the ordinance, eg. only trained personnel may use medical devices. If medical devices are used repeatedly, they must always function correctly and be sterile. For active medical devices MPBetreibV sets further framework conditions. These include the provision of a suitable infrastructure and the development of a documentation system.
For the Medical Device Operator Ordinance, important changes came into force in 2014 and 2017.

Medical Device Regulation (MDR) - and other EU regulations for medical devices
Previously, the Medical Device Regulation (Directive 93/42 / EWG) applied to medical devices and the Active Implantable Medical Device (Directive 90/385 / EWG) to active implantable medical devices.
Currently, a significantly higher documentation effort for manufacturers is emerging. This applies especially to products with increased risk. Market surveillance authorities can determine, according to MDR, whether products are legally compliant. If no EU declaration of conformity has been issued, the declaration is incomplete or technically not documented, the medical device can be described as officially non-compliant. If the manufacturer does not restore compliance within the time limits, the medical device may be prohibited from entering the market.
The Medical Device Regulation combines two independent medical device directives. In addition, MDR specifies specific requirements:
- For the content of the technical documentation. These are regulated in more detail in Annex 2 of the MDR.
- The identifiability of medical devices. Each product must have a unique device identification number (UDI).
- To the labeling of medical devices.
- To the new responsible person. Each manufacturer names a responsible person who has qualified specialist knowledge of the respective medical device.
- For the provision of data for EUDAMED. The European database for medical devices will be greatly expanded. EUDAMED will not only be accessible to state institutions, but also for manufacturers and the public.
- To clinical reviews and exams through post-market data. Clinical evaluations need to be updated using post-market data of the post-market surveillance.
- For high-risk products. As part of the MDR, "special notified bodies" are used for high-risk products. In the future, only these specially designated bodies will be able to carry out conformity assessments for high-risk products.
- To "special notified bodies". These may be required in the future to include expert panels through consultation / scrutiny procedures. These bodies would have to report any new application for conformity assessment for a high-risk product of this Medical Device Coordination Group.
- To products with hazardous substances. Medical devices containing carcinogenic, mutagenic or reproductive toxicants must meet higher requirements.
- To the classification of some products. Many products previously classified as Class IIb now meet the requirements of Class III. Software hardly falls into class I.
- To the reprocessing of disposable products. These have increased significantly.
The EN ISO 17664 standards series specifies the information manufacturers must provide for the reprocessing (cleaning, disinfection, and, if applicable, sterilization) of medical devices. The goal is safe reuse and minimization of infection risks.
The standards apply to reusable medical devices that must be cleaned, disinfected, and/or sterilized before reuse. Part 1 covers products contacting sterile body areas, mucous membranes, or compromised skin; Part 2 covers non-critical products with contact to intact skin or no patient contact.
Manufacturers must provide detailed, validated instructions for all reprocessing steps: pre-treatment, cleaning, disinfection, sterilization (if applicable), drying, inspection, packaging, storage, and transport.
Only validated reprocessing procedures ensure that medical devices are safely and effectively cleaned, disinfected, and/or sterilized. Validation is essential for compliance with the EN ISO 17664 standards series and regulatory requirements.
HygCen provides comprehensive consulting, testing, and validation of your reprocessing instructions according to EN ISO 17664-1 and -2. We support you from risk analysis to documentation and assist with audits and regulatory inquiries.