Jetzt Kontakt zu uns aufnehmen!
Sie erreichen uns unter folgender Telefonnummer:
+49 (0) 385 477 419 00




HygCen prüft Ihr Medizinprodukt auf biologische Unbedenklichkeit. Grundlage dafür ist die Biologische Beurteilung von Medizinprodukten gemäß
EN ISO 10993-1.
Medizinprodukte sind biologisch unbedenklich, wenn die eingesetzten Materialien mit dem Gewebe, den biologischen Zellen und den Körperflüssigkeiten der Patient*innen oder Anwender*innen wechselseitig verträglich sind.
Ihr Medizinprodukt darf bei korrekter Anwendung weder Patient*innen, Anwender*innen noch Dritte vermeidbaren Risiken aussetzen. Dafür verantwortlich sind Sie als Hersteller oder deren Bevollmächtigter.
Medizinprodukte müssen daher auf ihre Biokompatibilität untersucht werden. Das gilt für Neuzulassungen und Modifikationen bewährter Produkte.
Die Biokompatibilitätsprüfung Ihres Medizinprodukts ist gesetzlich vorgeschrieben, wenn es nah am oder im Körper getragen wird.

Ihr Produkt ist nicht dabei? Kontaktieren Sie uns!
Selbst wenn der Gesetzgeber für Ihr Produkt keine biologische Beurteilung verlangt, kann diese als wichtiges Verkaufsargument genutzt werden.
Wir führen wöchentlich Zytotoxizitätstests durch. Je nach Voraussetzung und Prüfumfang ist ein Biokompatibilitätstest Ihres Produkts innerhalb von 10 Tagen möglich. Bei umfangreichen Tests benötigen wir 4 bis 6 Wochen nach Produkteingang für die biologische Beurteilung.
Einen Pauschalpreis können wir Ihnen nicht nennen. Je nach Prüfaufwand und –umfang variieren die Kosten. Gerne erstellen wir Ihnen ein unverbindliches Angebot auf Basis konkreter Informationen von Ihrem Medizinprodukt.
Die EN ISO 10993-Normenreihe legt die Anforderungen und Prüfmethoden der biologischen Beurteilung von Medizinprodukten fest. Welche Prüfungen wir konkret für Ihr Medizinprodukt durchführen, ist abhängig von der Klassifizierung und Anwendung Ihres Medizinprodukts.
In welche Klasse Ihr Produkt fällt, ist abhängig von der Art und der Dauer seines Körperkontakts mit den Patient*innen. Diese Klassifizierung bestimmt, welche Prüfungen Ihr Produkt im Rahmen der biologischen Prüfung durchlaufen sollte, bevor Sie es auf den Markt bringen.
HygCen bietet alle biologischen Tests im Rahmen der EN ISO 10993 Reihe, die keine Tierversuche beinhalten, an. Wir testen dabei garantiert cruelty-free, also ohne Tierversuche. Dafür verwenden wir in vitro Testverfahren. Außerdem prüfen wir Ihr Medizinprodukt im Originalzustand. Das bedeutet, dass unsere Tests auch Prozessrückstände, Fremdsubstanzen oder Migrationen von Substanzen (z.B. aus dem Verpackungsmaterial) mitbewerten.
In vitro Prüfungen finden im Reagenzglas statt. Gemäß EN-ISO-10993-1 und -2 hat der Tierschutz im Prozess der Biokompatibilitätsprüfung eine hohe Priorität. Ihr Prüfplan zur biologischen Verträglichkeit von Medizinprodukten sollte sich daher besonders auf in vitro Testverfahren stützen. In vitro Testverfahren sind für Sie: kostengünstig, schnell und tierfreundlich.
In vivo Prüfungen finden in Tiermodellen statt. Diese Tests sind kosten- und zeitintensiv. Wenn Risiken nicht korrekt und vollständig über in vitro Tests nachgewiesen werden können, sind in vivo Tests notwendig. Für Medizinprodukte wie Implantate ist das relevant. Über in vivo Tests können die akute/subchronische/chronische Toxizität, die systemische Toxizität und Implantationen beurteilt werden.
HygCen prüft ohne Tierversuche!
Der Zytotoxizitätstest ist ein entscheidender Test im Rahmen der biologischen Beurteilung von Medizinprodukten. Der kostengünstige Test erfüllt eine Basisfunktion. Wird diese Prüfung nicht bestanden, sind i. d. R. keine weiteren Tests sinnvoll.
Testverfahren:
Der Zytotoxizitätstest umfasst mehrere in vitro Prüfmethoden. Primär unterscheiden sich diese Tests durch die Bestimmung des Endpunktes. Das prinzipielle Verfahren können Sie sich folgendermaßen vorstellen:
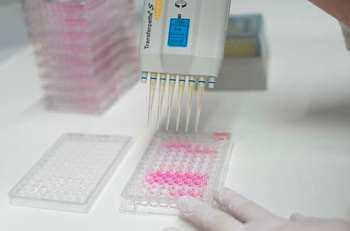
Alle Medizinprodukte, die durchgehend länger als 30 Tage angewendet werden, müssen auf Genotoxizität untersucht werden. Dieser Test muss unabhängig vom Zytotoxizitätstest angewendet werden.
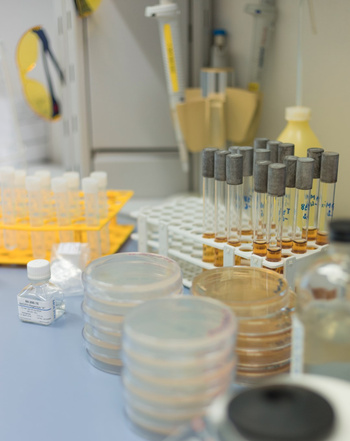
Testverfahren:
Genotoxizität wird durch in vitro Tests bestimmt. Die Prüfungen sind schnell, kostengünstig und relativ einfach durchführbar.
Der primäre Vorauswahltest ist das Ames Bacterial Reversion Assay. Für diesen Genotoxizitätstest werden spezielle Bakterienstämme benutzt. Diese Bakterienstämme können selbst kein Histidin mehr bilden und sind daher auf dessen Zuführung angewiesen. Generell besitzen die Bakterien aber noch die Gene, die für die Histidin-Herstellung notwendig sind. Durch Remutation können die Gene wieder aktiv werden.
Wenn Ihr Medizinprodukt mit dem Blutkreislauf in Kontakt kommt, muss es auf Hämokompatibilität untersucht werden. Primär testen wir dabei, ob Ihr Produkt Thromben und Embolien auslösen kann. Diese Verursacher von Schlaganfällen und Herzinfarkten müssen Sie unbedingt vor Markteintritt ausschließen. Neben der chemischen Zusammensetzung gilt es hier auch die physikalische Eigenschaft zu untersuchen. Die Oberflächenbeschaffenheit Ihres Medizinprodukts kann die Interaktion mit den verschiedenen Blutkomponenten stark beeinflussen.

Testverfahren:
Es gibt verschiedene kostengünstige in vitro Testverfahren für Hämokompatibilitätstests. In diesen Tests bestimmen wir die Hämolyse, Koagulation und Komplementaktivierung im menschlichen Blut.
Medizinprodukte, Hilfsmittel (Strümpfe und Verbände), u. a. müssen auf potenzielle Zellzerstörungen/Irritationen untersucht werden. Hat ihr Medizinprodukt den Zytotoxizitätstests bestanden, kann zusätzlich der Irritationstest sinnvoll sein. Dieser Provokationstest soll ausschließen, dass Ihr Medizinprodukt Kontaktallergien auslösen kann.
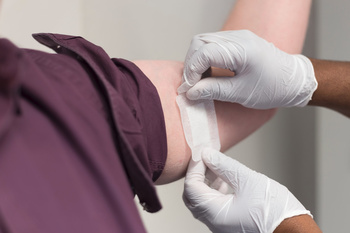
Testverfahren:
Die Irritationstests werden in der Regel in vivo durchgeführt. Das bedeutet, dass die Hautirritationstests am menschlichen Probanden durchgeführt werden, nachdem sie zuvor die Zytotoxizitätstest bestanden haben. In diesem Test wird Ihr Medizinprodukt oder Extrakte davon, direkt auf der Haut untersucht.
In diesem Test prüfen wir, ob Ihr Medizinprodukt frei von Endotoxinen und Pyrogenen ist. Pyrogene Reaktionen (Fieberreaktionen) werden meist durch bakterielle Verschmutzung hervorgerufen. Endotoxine sind die Fremdkörper, die das Fieber meist auslösen. Endotoxine oder auch Lipopolysaccharide sind Bestandteil der äußeren Hülle bestimmter Bakterien. Das menschliche Immunsystem kann Endotoxine aufspüren und mit Fieber reagieren.
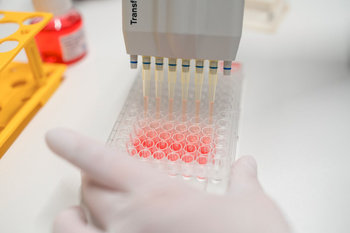
Testverfahren:
Die Endotoxin- und Pyrogentests führen wir in vitro durch.
Der MAT-Test (Monozyten Aktivierungstest) kann Endotoxine und andere pyrogene Substanzen nachweisen: z. B. Viruspyrogene, Pilzkomponenten, bakterielle Teichonsäuren u. a.
Die chemische Charakterisierung in der biologischen Beurteilung von Medizinprodukten nimmt an Bedeutung zu. Die Normen EN ISO 10993-1, -17 und -18 und vor allem deren Überarbeitungen in den zeigen diese Entwicklung. Entsprechend muss die chemische Zusammensetzung vor biologischen Prüfungen bekanntsein. Wenn bei der chemischen Charakterisierung des Medizinprodukts ein geeigneter toxikologischer Grenzwert verwendet wird, kann entschieden werden, ob die jeweilige biologische Prüfung notwendig ist. Zusammen mit in vitro‑Verfahren für biologische Endpunkte können auf diesem Wege vor allem Tierversuche (in vivo‑Verfahren) im besten Fall vermieden werden, was für Sie als Hersteller von Medizinprodukten finanzielle und zeitliche Vorteile bringt.
Die chemische Charakterisierung von Medizinprodukten ist ein Hauptbestandteil und vor allem einer der ersten Schritte der biologischen und toxikologischen Bewertung gemäß Normreihe EN ISO 10993. Dabei rücken komplexere chemisch-analytische Prüfungen in den Fokus, die auch geeignet sind, einen Gesamtüberblick an Substanzen, die im Medizinprodukt enthalten sind, in Form eines Screenings zu erhalten. Mittels geeigneter Techniken können diese zunächst unbekannten Substanzen identifiziert und ihr Gehalt im Medizinprodukt bestimmt werden.
Sie erreichen uns unter folgender Telefonnummer:
+49 (0) 385 477 419 00
Sie finden uns hier:
HygCen Germany GmbH
Bornhövedstrasse 78
19055 Schwerin